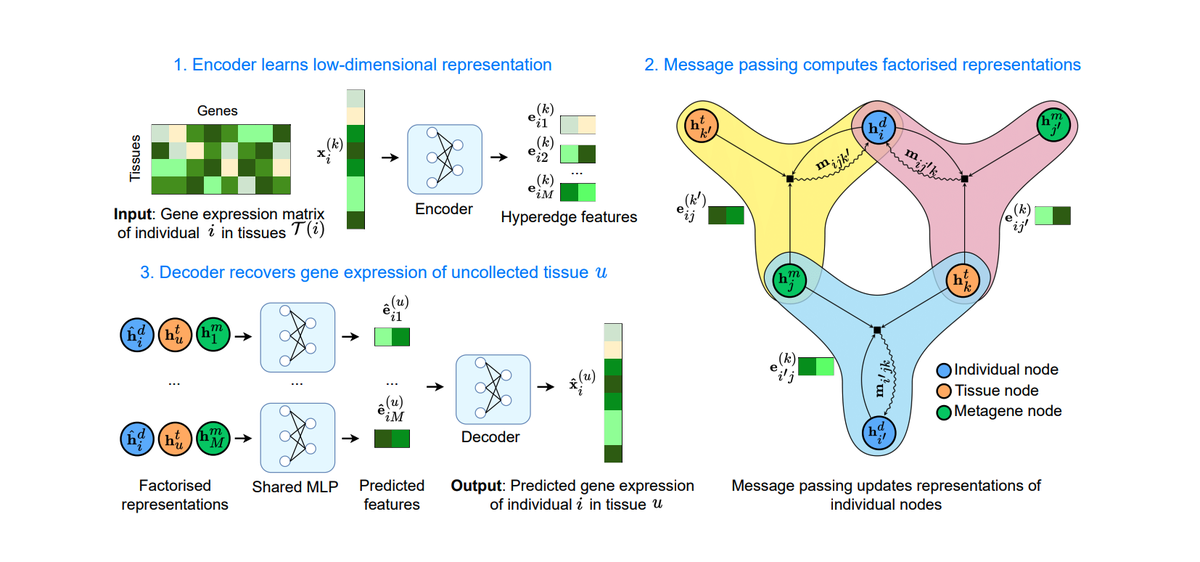
Abstract
Integrating gene expression across scales and tissues is crucial for understanding the biological mechanisms that drive disease and characterise homeostasis. However, traditional multi-tissue integration methods cannot handle uncollected tissues or rely on genotype information, which is subject to privacy concerns and often unavailable. To address these challenges, we present HYFA (Hypergraph Factorisation), a novel method for joint imputation of multi-tissue and cell-type gene expression. HYFA imputes tissue-specific gene expression via a specialised graph neural network operating on a hypergraph of individuals, metagenes, and tissues. HYFA is genotype- agnostic, supports a variable number of collected tissues per individual, and imposes strong inductive biases to leverage the shared regulatory architecture of tissues. In performance comparison on data from the Genotype Tissue Expression project, HYFA achieves superior performance over existing transcriptome imputation methods, especially when multiple reference tissues are available. Through transfer learning on a paired single-nucleus RNA-seq (snRNA-seq) dataset, we further show that HYFA can accurately resolve cell-type signatures from bulk gene expression, highlighting the method’s ability to leverage gene expression programs underlying cell-type identity, even in tissues that were never observed in the training set. Using Gene Set Enrichment Analysis, we find that the metagenes learned by HYFA capture information about known biological pathways. Notably, the HYFA-imputed dataset can be used to identify regulatory genetic variations (eQTLs), with substantial gains over the original incomplete dataset. Our framework can accelerate effective and scalable integration of tissue and cell-type gene expression biorepositories.